La farmacoeconomía de la terapia oncológica abre un amplio campo de investigación respecto de la descripción y análisis de sus costos, y de las consecuencias y resultados del tratamiento del cáncer para los pacientes, los sistemas sanitarios y la sociedad. La forma en que un sistema de patentes interactúa con el proceso de aprobación de medicamentos es un claro ejemplo de cómo pueden distorsionarse las decisiones de inversión respecto de la ejecución de ensayos clínicos destinados a combatir el cáncer. Si el objetivo básico de una política de medicamentos reside en garantizar la disponibilidad de tratamientos seguros, eficaces y de calidad, es importante poder asumir los gastos que estos originan por parte del sistema de salud más allá del costo de oportunidad que representan. Cuando los beneficios no son significativos, pero los precios resultan excesivos, la política de medicamentos entra en crisis. Lo que ocurre en el campo de la terapia oncológica demuestra que no estamos tan lejos de la misma.
El cáncer es la segunda causa de muerte en los EE.UU. después de las enfermedades del corazón, con unos 580.000 nuevos casos al año y un promedio de aproximadamente 1,600 muertes por día. A nivel mundial, el 60 por ciento de todas las muertes por cáncer se producen en países en desarrollo, donde los expertos sostienen que la incidencia de la enfermedad está creciendo rápidamente. Esto ha añadido urgencia a un debate activo y necesario respecto de si los esfuerzos para combatirlo son suficientes, y si no resulta estratégico repensar la escasa dedicación a la investigación en prevención. También es frecuente que en revistas científicas y periódicos que disponen de columnas sobre salud aparezcan artículos e historias respecto del comportamiento de la industria farmacéutica frente a las enfermedades oncológicas. Algunas noticias son de impacto positivo, resultado de nuevos descubrimientos terapéuticos o mejores logros asistenciales frente a problemas complejos de resolución. Por ejemplo mejor control u obtención de remisión en ciertos cánceres. La contracara la conforman noticias de sesgo negativo, generalmente económicas, vinculadas al peso que la escalada de precios de las nuevas drogas oncológicas, especialmente las biológicas, ejerce sobre los financiadores de la salud y los propios pacientes.
La industria innovadora del medicamento, conocida como «Big Pharma» e integrada por las grandes empresas transnacionales, se encuentra inmersa en una frenética carrera por obtener primacía en los frentes estratégicos de producción y desarrollo de drogas biotecnológicas tanto oncológicas como “huérfanas”, en mejores oportunidades de negocios a través de una corriente de adquisiciones con concentración del mercado, y principalmente en la obtención de beneficios financieros y accionarios provenientes de fondos de terceros no vinculados al sector. El principal inconveniente que ofrece esta competencia es el precio con que cada una de las nuevas drogas ingresa al mercado sanitario. Ya no se está pagando ese precio por la innovación, ni por menos efectos colaterales o mejor nivel de salud. Se lo está pagando en función de un modelo de negocios basado en la obtención de beneficios de muy alta rentabilidad.
Hay casi una docena de drogas oncológicas cuyos precios exceden los U$ 100.000 al año y se administran a pacientes con enfermedad en grado avanzado. Pagar esa cantidad de dinero quizás no tenga mucho sentido, dado el escaso beneficio terapéutico que ofrecen, valorado en cantidad o calidad de vida. El astronómico precio de los nuevos medicamentos representa los peores excesos de la industria sobre los sistemas de salud, que se limitan a ejercer débiles controles sobre el poder de fijación de precios y a priorizar pequeñas ganancias en salud por sobre la realidad de los mayores costos. Ciertamente los ciudadanos estarían de acuerdo en que sería mejor prevenir el cáncer, si esto fuera posible, en lugar de seguir asumiendo tratamientos cada vez más complejos y costosos. Sin embargo, los incentivos económicos y las cuestiones regulatorias alientan a los investigadores de la industria y a sus directivos a centrarse prioritariamente en mejoras de tratamiento en lugar de desarrollos de terapéuticas para mejorar la prevención.
La farmacoeconomía de la terapia oncológica abre un amplio campo de investigación respecto de la descripción y análisis de sus costos, y de las consecuencias y resultados del tratamiento del cáncer para los pacientes, los sistemas sanitarios y la sociedad. La forma en que un sistema de patentes interactúa con el proceso de aprobación de medicamentos es un claro ejemplo de cómo pueden distorsionarse las decisiones de inversión respecto de la ejecución de ensayos clínicos destinados a combatir el cáncer. Si el objetivo básico de una política de medicamentos reside en garantizar la disponibilidad de tratamientos seguros, eficaces y de calidad, es importante poder asumir los gastos que estos originan por parte del sistema de salud más allá del costo de oportunidad que representan. Cuando los beneficios no son significativos, pero los precios resultan excesivos, la política de medicamentos entra en crisis. Lo que ocurre en el campo de la terapia oncológica demuestra que no estamos tan lejos de la misma.
Encontrar un equilibrio entre costos y consecuencias es crucial a cualquier esquema económico. Una forma de maximizar la salud de la población es poner un cierto equilibrio entre inversión y ganancias, costo y calidad y entre necesidades y deseos de los pacientes y de la sociedad, reconociendo la imposibilidad de satisfacerlos en forma diferente o indistinta. Hay más dinero para inversión en I+D sobre drogas que permitan extender la vida de pacientes en etapas tardías de la enfermedad por un par de semanas o meses, que en tratamientos que impidan que la enfermedad se desarrolle. Y son escasas las investigaciones en búsqueda de drogas que posean algún mérito clínico significativo para tratar la enfermedad en etapas tempranas, o vacunas que la prevengan.
La alternativa entre tratamiento curativo, paliativo o prevención y AVAC. El gran dilema
Hace pocos días terminé de leer datos muy interesantes respecto de un estudio realizado por investigadores del Instituto de Tecnología de Massachusetts (MIT) y la Universidad de Chicago, publicado en el diario New York Times (NYT) bajo el título ¿Why Preventing Cancer Is Not the Priority in Drug Development?. Me surgió una pregunta inmediata: ¿Por qué en una guerra de casi 50 años contra el cáncer no se produjo ningún resultado en el hallazgo de un medicamento o nuevas terapias que favorezca su prevención, o al menos una respuesta terapéutica altamente efectiva? Parafraseando a Hamlet: He aquí el dilema.
Una primera conclusión del artículo, basado en una investigación efectuada por dos docentes de economía, Heidi Williams, profesora de la Fundación MacArthur y Eric Budish de la Universidad de Chicago, junto a Ben Roin, asistente de innovación tecnológica, es que existen una serie de razones por las cuales la industria farmacéutica no ha priorizado en forma significativa la investigación aplicada a la prevención del cáncer, como sí lo ha hecho con otras drogas novedosas. Los autores comentan que lograr acelerar la disponibilidad de uso de los medicamentos para tratar enfermedades graves resulta de interés de todos los actores del sistema de salud, sean pacientes, médicos, instituciones asistenciales y compañías farmacéuticas. Sobre todo cuando estas drogas constituyen la primera alternativa terapéutica posible y poseen evidentes ventajas por sobre los tratamientos preexistentes. Para disponer de mayor celeridad sobre la aprobación de nuevos compuestos, la Food and Drug Administration de EEUU (FDA) ha generado cuatro estrategias que apuntan a favorecerla:
- Fast Track
- Breakthrough
- Aprobación acelerada
- Revisión prioritaria
Por Fast Track se conoce un proceso diseñado para facilitar el desarrollo de ciertos medicamentos, a partir de acelerar su revisión para permitir el tratamiento de enfermedades graves con alto riesgo de vida. Debe ser solicitado por la compañía farmacéutica, pudiendo ser iniciada en cualquier momento durante las fases de desarrollo del fármaco. LA FDA revisa la solicitud y toma una decisión dentro de los sesenta (60) días en función de si el fármaco completa una necesidad médica insatisfecha para pacientes en estado grave. Toda vez que un fármaco recibe la designación Fast Track, es necesario establecer una comunicación frecuente entre la FDA y la compañía farmacéutica innovadora a lo largo de todo el proceso de desarrollo del fármaco y durante la etapa IV de farmacovigilancia. La frecuencia de comunicación asegura que toda pregunta, inconveniente o problema vital se resuelva rápidamente.
Por su parte, el Breakthrough permite acelerar el desarrollo y la revisión de medicamentos que pueden demostrar una mejora sustancial respecto de una terapia ya disponible. Para determinar si la mejora con respecto a esta es sustancial, el criterio depende tanto de la magnitud del efecto terapéutico como su duración y la importancia de la evolución clínica observada. En general, la evidencia clínica preliminar debe mostrar una clara ventaja respecto del tratamiento disponible.
La Aprobación acelerada data de 2012. Mediante la aprobación por parte del Congreso de EEUU de la Ley de Innovación de Seguridad de Medicamentos y Alimentos (FDASIA), se permite a la FDA autorizar el ingreso al mercado sanitario de ciertas drogas destinadas a tratar enfermedades graves y que llenen una necesidad médica no satisfecha a través de criterios indirectos de valoración. Un criterio indirecto utilizado para la aprobación acelerada es un marcador – una medición de laboratorio, imagen radiográfica/tomográfica/por RNM o PET, un signo físico o cualquier otra medida que pueda predecir el beneficio clínico, aunque no sea medida exacta del mismo, como un efecto positivo sobre la morbilidad y mortalidad irreversible (IMM).
Finalmente, por Revisión prioritaria se entiende que la FDA puede tomar decisión respecto de una solicitud de aprobación dentro de los 6 meses de presentada, en comparación con los 10 meses que posee la revisión estándar. Una revisión prioritaria dirigirá atención y recursos humanos a la evaluación de solicitudes de medicamentos que, de aprobarse, traerán importantes mejoras en la seguridad o eficacia del tratamiento, el diagnóstico o bien la prevención de enfermedades graves, en comparación con las terapias estándar. La designación de un medicamento como «prioridad» bajo ningún concepto altera la norma médica o científica necesaria para su aprobación o la calidad de las pruebas de evidencia requeridas.
Estos procedimientos definen la razón de elegibilidad de las Big Pharma respecto de innovar en drogas que comprenden determinada banda terapéutica, por ejemplo el campo de las oncológicas. Resulta económicamente más rentable centrarse en el rápido desarrollo e ingreso al mercado de fármacos que prolonguen el tiempo de sobrevida – aunque sea limitado – más que en mejorar la calidad de vida, por ejemplo ayudando a la prevención de la enfermedad.
La segunda conclusión es que si bien se suele sostener que la I + D en prevención y tratamiento del cáncer en etapas tempranas constituye un hecho socialmente muy valioso, la misma sociedad proporciona a las empresas privadas – tal vez inadvertidamente – pocos incentivos para llevar adelante este tipo de investigaciones. ¿Es preferible tratar la enfermedad que adelantarse a que aparezca? ¿Dónde está el nudo del problema? Está en que ciertas cuestiones como el retraso en la aprobación y autorización de comercialización de una determinada droga, o bien el tiempo que transcurre entre la recepción de la solicitud de la patente y la aprobación de la FDA, incentivan el desarrollo de fármacos que puedan ser aprobados rápidamente, aunque en forma paradójica posean beneficios de sobrevida comparativamente acotados. Con un tamaño de muestra suficientemente grande, una prolongación estadísticamente significativa de la supervivencia global puede demostrarse incluso si el fármaco mejora la duración de la vida sólo por unos pocos días o semanas. Las drogas también pueden ser aprobadas en base a criterios indirectos, por ejemplo reducción de la masa tumoral o descenso en los biomarcadores, sin pruebas evidentes de que los pacientes se beneficien de una mejora en la supervivencia.
La tercera conclusión radica en que la forma en que el sistema de patentes interactúa con el proceso de aprobación de medicamentos de la FDA distorsiona las decisiones sobre diferentes tipos de ensayos clínicos específicamente destinados a resolver el problema del cáncer. Hay más recursos económicos para invertir en el desarrollo de medicamentos que extiendan la vida de los pacientes oncológicos por un par de meses y recuperar la inversión de su descubrimiento, que en medicamentos que efectivamente impidan o bloqueen su aparición.
La cuarta está basada en la esperanza que incorporan los monoclonales, muchos aún en fase de investigación de efectividad. Sostienen que “La industria quiere que se siga teniendo la esperanza en el largo plazo de encontrar algo que revolucione el tratamiento del cáncer, mientras los pacientes mueren por miles en el corto y mediano plazo”. Así llegan a demostrar que para asegurar la aprobación de la patente por la F.D.A de un futuro medicamento superstar, “las compañías farmacéuticas corren contra el tiempo para demostrar en Fases II y III que el producto es más seguro y eficaz”. La pregunta es frente a que droga alternativa lo hacen, o si es solo contra placebo.
El sistema de patentes ofrece, tal vez inadvertidamente, muy poco incentivo a las empresas privadas que se dedican a investigaciones de largo plazo. En el caso de las drogas oncológicas, cuanto más rápido sea posible completar los estudios, mayor será el tiempo de protección de patente sobre la molécula hasta que se produzca su vencimiento, período durante el cual los márgenes del beneficio económico por posición de monopolio resultaran más elevados.
Siguiendo con el mismo estudio. También se destaca el número de ensayos de drogas oncológicas efectuado antes de la aparición de las biotecnológicas monoclonales. Entre 1973 y 2011 hubo alrededor de 12.000 investigaciones en Fase II y III para pacientes en etapas relativamente tardías de evolución de la enfermedad, con una probabilidad del 90% de morir en cinco años. Otras 6.000 se centraron en pacientes cuya probabilidad era solo del 30% de morir. Y más de 17.000 ensayos comprendieron pacientes con la probabilidad más baja de supervivencia (cáncer recurrente o multimetastásico). Lo particular es que sólo 500 investigaciones estuvieron vinculadas a la prevención del cáncer, factor que otorgaría los beneficios más largos en cuanto a supervivencia. Este sesgo hacia estudios centrados en pacientes con menores oportunidades de sobrevida ha sido frecuente con medicamentos cuya I+D proviene de financiación privada en lugar de pública.
Por lo general, los ensayos clínicos necesarios para que la F.D.A. apruebe un medicamento requieren una duración de varios años. Según datos provistos por la industria farmacéutica innovadora, 58% del costo de desarrollar un medicamento se consume en los ensayos clínicos de fase III. El tiempo promedio de aprobación de una droga oncológica desde el comienzo del testeo clínico es aproximadamente de 8 años. Aunque la patente virtual pueda alcanzar 20 años (o más si se lo pasa a evergreen), un medicamento tipo llega al mercado con cerca de 12,5 años reales de vida restante bajo protección. La Ley de Cuidado de Salud Asequible del Presidente Obama incluye una disposición que otorga 12 años de exclusividad en el mercado a partir de la aprobación por la FDA – medio año menos de los típicos 12,5 años restantes en una patente – pero sólo a los medicamentos biotecnológicos.
Quienes más procuran tener algún tipo de control sobre el tiempo entre la recepción de una patente y su aprobación para comercialización por la FDA son precisamente las BigPharma innovadoras. Mediante el estudio de pacientes en los cuales garantizar seguridad y eficacia puede tener rápido trámite, logran reducir este retraso. La FDA ha comenzado hace tiempo a aprobar los medicamentos sobre la base de la evidencia de beneficio y seguridad clínica, sin un claro umbral de requerimientos en Fase III respecto del beneficio necesario alcanzado para justificar dicha aprobación.
La particularidad es que el desarrollo de fármacos para tratar el cáncer en etapa avanzada suele ser mucho más rápido que para una etapa temprana o de prevención, porque la enfermedad en fase tardía resulta ser muy agresiva y rápidamente progresiva y lo que se necesita es ofrecer tanto seguridad como cierta eficacia. Esto permite a las empresas disponer de resultados de los ensayos clínicos con mayor celeridad. Incluso si sólo consisten en una pequeña mejora clínica cuantitativa y no cualitativa en cuanto a supervivencia, aun comparada con otros fármacos ya existentes en el mercado. Ocurre también que algunos compuestos no completan los estudios, simplemente porque el tiempo de protección disponible que otorga la patente para recuperar el costo del desarrollo de la molécula resulta demasiado corto.
De allí que cada vez más, las grandes compañías farmacéuticas apuestan a encontrar nuevas moléculas contra el cáncer que cuestan miles de millones de dólares de desarrollo y se venden por miles de dólares por dosis. Por ejemplo, en el año 2010 diez medicamentos oncológicos autorizados se transformaron en blockbusters, al haber alcanzado – según la Consultora Campbell Alliance – el billón de dólares en ventas. En los diez años anteriores, solo dos productos habían podido lograrlo.
Pero una cosa es el precio y otra muy diferente el beneficio. Por lo general, las drogas oncológicas más novedosas y caras no aportan resultados que permitan prolongar la vida en forma dramática a los pacientes. Especialmente ocurre con algunos biotecnológicos que exhiben datos estadísticamente significativos pero clínicamente de escaso beneficio relevante. Estos beneficios en salud se representan en función de Años de Vida Ajustados por Calidad (AVAC o QUALY´s) y no por cantidad de tiempo, obtenidos a partir del momento en que se inicia el tratamiento. La medida del AVAC expresa el número de años de vida que se añadirían debido a determinada intervención terapéutica, ajustados por calidad de vida. Es un indicador clave que se utiliza comúnmente en la evaluación de la relación calidad – precio de un medicamento o dispositivo, y refleja la mejora cualitativa de la vida en forma más específica y menos subjetiva que la cantidad de vida que puede ofrecerse bajo determinadas condiciones.
Un AVAC con valor 1 es el equivalente a un año de vida vivido sin ninguna forma de enfermedad Por su parte, un valor de 0,5 indica una calidad de vida 50% de lo normal. Por lo tanto, una ganancia de 1 año completo a una Q de 0,5 equivale a una ganancia de medio año en perfecto estado de salud (AVAC = 1 × 0,5 = 0,5 años). Cero equivale a estar muerto, ya que no es posible acumular AVAC después de morir.
Los beneficios de una nueva droga se definen como el número de AVAC adicionales experimentados por los pacientes a quienes se administró la misma, comparado con una opción de tratamiento preexistente o contra placebo. Una vez que se determina el AVAC, el siguiente paso es calcular la relación costo-efectividad incremental (ICER). Supongamos que para una afección potencialmente mortal grave, el tratamiento estándar cuesta U$3000 y ofrece 1 año de vida con una calidad prevista de 0,4. El AVAC asociado a este tratamiento es, por lo tanto, de 0,4 años. Ahora supongamos que un nuevo tratamiento que cuesta U$50.000 ofrece 2 años de vida con un Q de 0,7. El AVAC en este estado es de 2 × 0,7 = 1,4 años. El ICER se calcula tomando la diferencia de costos entre los 2 tratamientos, dividido por la diferencia en los AVAC entre los dos tratamientos:
ICER = (U$ 50,000- U$ 3000) / (1,4 – 0,4) = $ 47,000 por AVAC ganado.
El mayor inconveniente de calcular costo/efectividad en oncología es respecto de drogas que no parece tener mucho sentido administrar, dado el elevado costo que poseen y los pequeños beneficios que agregan. Por ejemplo, algunas novedades biotecnológicas pueden llegar a costar más de U$ 10.000 por mes, pero proveen una media de sobrevida adicional que va desde pocas semanas a menos de un mes, asociado a una elevada toxicidad sustancial y efectos adversos.
Chambers y colaboradores (2014) revisaron efectividad (en términos de QUALY/AVAC) y razón coste-efectividad de una serie de fármacos destinados a tratar diferentes enfermedades consideradas graves, incluidas las oncológicas, que fueron aprobados por la FDA entre 1999 y 2011. Demostraron que la gran mayoría logro beneficios modestos en la salud de los pacientes frente a lo que se lograban con otras drogas ya existentes en el mercado sanitario. Las estimaciones respecto de los beneficios adicionales de salud (medidos en AVAC) y el aumento de costos se efectuaron sobre cincuenta y ocho nuevas drogas y cuarenta y cuatro fármacos tradicionales. De los nuevos medicamentos analizados, un 32% no ofreció ningún beneficio adicional. De los que sí lo hicieron, un tercio aportó menos de 0,1 AVAC incremental (equivalente a 5 semanas de supervivencia ajustada por calidad) en tanto los dos tercios restantes otorgaron menos de 0,3 AVAC (15 semanas). (Ver Cuadro 1)
En el mismo estudio, catorce fármacos de última generación y dos tradicionales mostraron poco más de 0.5 AVAC de beneficio (25 semanas de supervivencia ajustada por calidad), y el cálculo de costo/efectividad resultaba elevado. Dos drogas registraban ratios de hasta U$
50.000/AVAC, tres entre U$ 50.000 y 100.000/AVAC y en seis eran superiores a U$ 250.000/AVAC. Un ejemplo era el imatinib (Gleevec®), administrable por vía oral para tratar la Leucemia mieloide crónica con alta eficacia. Chambers señaló que ésta posee el tercer mayor costo incremental ($ 151.476) y el segundo como ganancia de AVAC (4,1 AVAC), con una relación costo-efectividad incremental de $ 36.921 por AVAC. Como dato de referencia, un umbral entre $50.000 a $ 100.000 por AVAC se justifica en EEUU como definición de intervención sanitaria rentable, basado en la diálisis. Para el Reino Unido y Canadá, el umbral de ICER deseado para la aprobación de tratamientos para el cáncer es por lo general menor de $ 50.000 por AVAC.
Cuadro 1. Drogas oncológicas elegidas por el mayor nivel de ganancias en AVAC respecto de terapias preexistentes
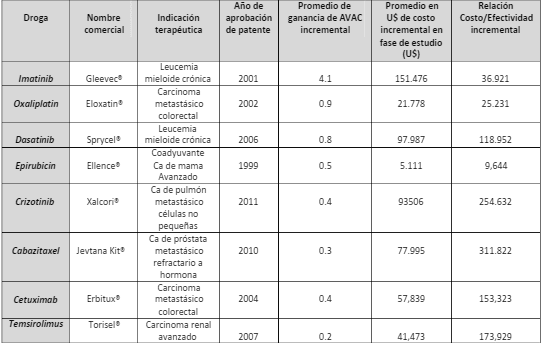
Fuente: Tomado de Chambers,J; Health Affairs, 33, no.10 (2014):1751-1760
Por lo tanto, si bien pueden ofrecer mayores ganancias de bienestar en términos incrementales respecto de los AVAC (0.183 versus 0,002 AVAC), muchas drogas presentan mayores costos adicionales incrementales (U$12.238 versus U$784). El 26% de los medicamentos bajo análisis presentaron ratios de costo/efectividad incremental muy superiores a los U$150,000 por AVAC. Este dato señala la necesidad de plantear un urgente debate respecto de cuál debiera ser el umbral de costo -efectividad que como sociedad se está dispuesto a pagar frente a la escalada de precios de las nuevas drogas.
Si bien en los últimos veinte años los investigadores han logrado descubrir un mayor número de secretos respecto de mutagénesis, tumorigénesis, expresión génica y proteica, sistemas de mensajería, angiogénesis y resistencia al tratamiento, la terapéutica aún resulta insuficiente para evitar un desenlace fatal. De acuerdo a una publicación de Siddiqui & col en Mayo Clinic Proceedings, al ser limitada la duración del efecto terapéutico de la mayoría de los medicamentos oncológicos, cuando un tratamiento deja de ser efectivo, es común que al paciente se le administre otra nueva droga hasta que todas las opciones posibles fracasen. Por ejemplo, si hay cuatro drogas aprobadas para tratar un cáncer incurable en particular, no existe posibilidad de obtener precios bajos, ya que todas serán aplicadas en alguna etapa de la evolución de la enfermedad, independientemente de la respuesta. Por lo común, los beneficios terapéuticos tienen una duración breve si se la mide en semanas o meses, luego de lo cual el tumor comienza a no responder a la medicación. Frente a esta situación, los médicos no suelen elegir la opción más costo/efectiva sino que solo deciden el momento oportuno para utilizar cada opción terapéutica. Por tal razón, no existe competencia por efectividad significativa entre las drogas oncológicas –muchas de ellas experimentales – que permita reducir sus precios, sino todo lo contrario. A la inversa de lo que ocurre con patologías menos complejas y curables, donde las compañías farmacéuticas han sido exitosas en desarrollar productos eficaces con menores efectos adversos y mejores precios.
Algo similar ocurre con otras enfermedades crónicas incurables o altamente discapacitantes frente a cada innovación terapéutica que se aprueba, como ocurre con muchas de las poco frecuentes. La magnitud económica del problema resulta más complejo en oncología. El impacto social de la enfermedad y la gravedad de su diagnóstico coloca a pacientes y médicos frente a la necesidad imperiosa de asumir los altísimos costos de tratamiento sin medir demasiado los resultados. Por ejemplo en términos de AVAC ganados.
Terapia oncológica y precios. Un camino donde se cruza la ciencia, la economía y la bioética
La Oncología, especialidad que fuera considerada un nicho de mercado hace algunos años atrás, logro alcanzar un volumen de negocios de U$41 billones en 2008, si se lo compara con los U$24 de 2004. A partir de este dato, los analistas del mercado de medicamentos de Estados Unidos han venido sosteniendo lo que definen como «financial toxicity«. Muchos de los nuevos fármacos oncológicos ingresan al mercado con un precio de más de U$ 100,000 al año en Estados Unidos. No reemplazan a otros ya existentes en el mercado, sino que se agregan a la lista aunque sin añadir mayor calidad ni cantidad de vida en forma significativa. El problema es que si se calcula el costo de un medicamento en $ 120,000, y la siguiente innovación no va a costar menos que eso, la necesidad de financiamiento pasará a ser mínimamente de un cuarto de millón de dólares superior. Esta disparada de precios supera ampliamente los supuestos beneficios terapéuticos de estas drogas y señala una enorme brecha entre su precio final y el costo real de producción. Investigadores británicos sostienen que el precio de cinco drogas oncológicas resulta ser 600 veces mayor de lo que cuesta producirlas. Por ejemplo, el análisis de las cifras de costo real de producción para la provisión durante un año del mencionado imatinib (Gleevec®) es de U$ 159. Pero su precio de mercado es de U$ 106.322/año en los EE.UU y de U$ 31.867/año en el Reino Unido, mientras que en Argentina oscila entre U$ 39.434 y U$ 25.442/año. Sin palabras.
Todo precio final de un medicamento también se considera función de la población a quien esta direccionado, de la vida media de la patente y del rendimiento de la inversión proyectada. Pero el insólito valor de venta que tienen ciertas innovaciones oncológicas no guarda relación directa con ninguna de estas variables. El argumento más utilizado por la industria se relaciona con los costos de trasladar los hallazgos de laboratorio al desarrollo de la totalidad de los estudios reglamentarios (las tres fases de ensayo clínico) que se requieren para lograr la aprobación de comercialización por parte de la FDA o la AEM. Lo cierto y demostrado es que esos costos no siempre provienen de la propia industria privada, sino del sector público y de los Institutos públicos sanitarios que apoyan la investigación.
Hay una variable que si es vinculante, y tiene que ver con la incurabilidad de muchos cánceres. Los pacientes son incorporados por los médicos a protocolos de tratamiento de cada nuevo producto autorizado (en forma secuencial o combinada) con el objetivo de investigar resultados clínicos y a la vez intentar prolongar su tiempo de vida. Esto crea, de hecho, una condición de monopolio virtual, ya que si bien no hay una eliminación automática de las drogas presentes en el mercado, estas pasan a ser visualizadas como subestandar de menor valor terapéutico y sin capacidad de generar competencia por precio. Como ocurre bajo esta condición de monopolio, los fármacos que efectivamente prolongan la supervivencia de los pacientes incurables – incluso por unas pocas semanas – pueden admitir cualquier precio que el mercado o los financiadores puedan soportar. La evidencia de esto queda representada por la inelasticidad/precio (el cambio porcentual en el uso asociado de un medicamento vinculado a un aumento porcentual en su precio) que poseen estas drogas altamente específicas.
Sea por la propia naturaleza de la enfermedad y lo grave de su diagnóstico, pacientes y médicos condicionan a los financiadores a hacerse cargo del alto precio de los nuevos tratamientos. Incluso frente a muy pequeñas mejoras marginales en la costo/efectividad incremental de las alternativas terapéuticas. Además toda nueva droga, independientemente de su costo y supuesta función innovadora, siempre tendrá la alternativa de ser judicializada en su provisión (vía recurso de amparo o tutela) en caso que su financiación por parte de un organismo asegurador no sea aceptada o bien se dilate temporalmente. Por lo tanto, los medicamentos oncológicos, al igual que los utilizados para ciertas enfermedades poco frecuentes, representan una condición de monopolio de uso y de precio. Bien por la complejidad biológica del cáncer y su nivel de respuesta frente al tratamiento químico o biotecnológico. O en base a su relación con el sistema de salud, con la autonomía médica y con la falta de regulación de uso en base a guías y protocolos, situaciones que proveen incentivos para administrar más quimioterapia sin tomar muchas veces en cuenta consideraciones de eficiencia y efectividad económicas y clínicas y efectos adversos.
El paso de la farmacología química a la biológica supone un giro copernicano en la forma de tratar el cáncer. Y la relación entre el valor clínico de una droga oncológica y su precio resulta diferente según el tipo de la misma y el área terapéutica en la cual sea utilizada. El BMJ publicó en 2015 un artículo de Allen Shaughnessy bajo el título “Monoclonal antibodies: magic bullets with a hefty price tag” en el que el autor realiza un análisis certero de la situación actual de estos productos farmacéuticos. Prevé que las ventas de los anticuerpos monoclonales llegarán a más de U$ 160 billones en los EE.UU. en los próximos años. ¿Cuánto cuesta entonces un mes de vida de una persona? ¿Y llegar a seis meses de vida? ¿Puede la supervivencia de un enfermo tener un precio impagable? Esta serie de preguntas clave constituye el dilema de los principales reguladores sanitarios en el Siglo XXI, sea la FDA o la EMA, frente a medicamentos con precios tan elevados.
Si bien hay nuevos desarrollos terapéuticos que impactan sobre la supervivencia en forma significativa, productos extraordinariamente caros brindan apenas algunas semanas o meses de vida. Muchos de ellos ingresaron en forma muy rápida al mercado sanitario, debiendo ser retirados meses después que se publicaran estudios acerca de su efectividad pero también de sus efectos adversos. Ciertos monoclonales que se posicionaron con determinada indicación terapéutica luego, al encontrársele otra, fueron retirados en forma oportunística del mercado sanitario por el propio laboratorio. Es el caso de Sanofi – Genzyme, que cambio el nombre comercial de uno de sus productos de idéntica composición y lo reingreso nuevamente con un precio altísimo para competir frente a otros rivales en diferente banda terapéutica. Esto ocurrió con el alemtuzumab, indicado primariamente para la Leucemia linfocítica crónica y más recientemente para la Esclerosis Múltiple. En el primer caso se lo conoce comercialmente como Campath®, administrable durante 12 semanas a un precio de U$ 1.930 por dosis. El costo total de tratamiento alcanza los U$60. 000/año.
Los pacientes con EM necesitan una fracción de la dosis utilizada en la Leucemia (dos aplicaciones por año), por lo que bajo ese esquema de precios, la segunda variante del alemtuzumab bajo en nombre comercial de Lemtrada® sólo costaría U$ 6.000 por año. Pero Sanofi ha confirmado que un ciclo completo de tratamiento tendrá un costo de aproximadamente U$ 95,000 en Alemania, donde el producto se ha puesto en marcha inicialmente. El costo por frasco de Lemtrada® resulta cercano a los U$11.700 y los pacientes suelen tratarse con ocho aplicaciones durante un período de dos años. Sus competidores en esa banda terapéutica son el natalizumab (Tysabri®) de Biogen Idec y el fingolimod (Gilenya®) de Novartis. Lo paradójico es que esta última droga, que se administra por vía oral, tiene precios mucho más elevados (US2.940/28 comprimidos) que el Campath®, al menos en la dosis utilizada por los pacientes, pero no que el Lemtrada® que es de uso intravenoso. Por su parte el Tysabri® de uso oral cuesta $ 55.000 por año, más cercano al precio del Campath® pero también por debajo del de Lemtrada®.
Sanofi – Genentech retiro el Campath® de la mayor parte de Europa y de EEUU, y solo lo mantiene en 50 países. La pregunta es cómo este laboratorio puede distanciar al Lemtrada® del Campath® – si se trata de la misma droga – con el fin de obtener un precio más alto de las dosis para la EM, y al mismo tiempo evitar el uso del Campath® más barato retirándolo del mercado sanitario. Y como puede justificar pasar de U$2.000 a U$12.000 en el precio de la misma dosis de alemtuzumab, con un aumento del 600%. ¿Cuál es el límite entonces entre lo económico, lo científico y lo bioético?
Ejemplos de ausencia de límites sobran. El cáncer de mama posee múltiples opciones de tratamiento que han permitido ampliar la supervivencia en años, por ejemplo en tumores con HER2. En este caso, la altura de la vara de precios puede resultar mayor que en el cáncer de páncreas, donde la media de supervivencia es sólo de seis meses. Veamos algunos casos. El bevacizumab (Avastin®) de Roche fue aprobado primero para el tratamiento del cáncer colorrectal en el año 2004 y posteriormente para el de mama metastásico en el 2008. Los resultados de dos ensayos aleatorios posteriores sobre esta última enfermedad, efectuados en 2009, demostraron que los pacientes si bien experimentan una ganancia estadísticamente significativa en la «supervivencia libre de progresión» (que mide el período de tiempo en el que el cáncer está bajo control), presentan pequeñas diferencias respecto de la supervivencia, no estadísticamente significativas. Pero es fortísima la disparidad entre el valor agregado y el precio, que no resulta sostenible. Un trabajo del BMJ calcula que en base al precio corriente de administrar bevacizumab (£1,848.80/mes para pacientes de 70 kg) y el tiempo promedio de tratamiento requerido (aproximadamente 10 meses), el costo total de tratamiento resulta cercano a las £18,500/paciente. Otro estudio, publicado por el New England Journal of Medicine, demostró que la droga extendía la vida por 4.7 meses (20.3 meses versus 15.6 meses) pero a un costo de U$42,800 a U$55,000. Un tercero, efectuado por Goldstein & col señala que dicha droga, utilizada como tratamiento de primera línea, provee un adicional de solo 0.10 AVAC (5 semanas de sobrevida) a un costo de $59,361. Esto implica una costo/efectividad incremental de U$571,240 por AVAC. Por tal motivo, el National Institute for Health and Care Excellence (NICE) del Reino Unido descarto financiar el bevacizumab, al estimar que a ese costo solo agregaba mínimos beneficios clínicos. En el mismo sentido, la FDA revocó la autorización para su utilización en cáncer de mama en 2011. Sin embargo, un panel de expertos convocado posteriormente por la National Comprehensive Cancer Network (2010) – un consorcio de principales centros de cáncer de EEUU – votó en contra de la eliminación del bevacizumab de la lista de medicamentos indicados como para el cáncer de mama avanzado.
Otros ejemplos comparados de uso múltiple y efectividad diversa pueden encontrarse en el análisis del ramicirumab (Cyramza®) de Eli Lilly, que fuera aprobado por la FDA en Abril de 2014. Posee similar acción que otros inhibidores de la angiogénesis como el ya mencionado bevacizumab (Avastin) y el ziv-aflibercept (Zaltrap®) de Sanofi en tratamiento combinado con FOLFIRI (ácido folínico, fluorouracilo e irinotecan). El Cyramza® fue presentado comercialmente como una estrategia novedosa para el tratamiento del cáncer gástrico avanzado, donde el Avastin® no resultaba eficaz. Otorgaba un 37% de supervivencia global media, de 5,2 contra 3,8 meses para placebo (1.4 de diferencia). Además mejoraba la supervivencia libre de progresión (SFP) de 1.3 a 2.1 meses (0.8 de diferencia). Posteriormente se lo indicó para tratar el cáncer de colon metastásico, donde competía contra buenos resultados del bevacizumab, aunque duplicaba el precio. En este caso la supervivencia global media de los tratados con ramicirumab fue de 13,3 meses, contra 11,7 meses (1,6 mes de diferencia) de los pacientes tratados con placebo más FOLFIRI. Posteriormente, la FDA autorizo su uso en el cáncer de pulmón no microcítico metastásico en forma combinada con docetaxel. Su efecto fue aumentar la mediana de supervivencia global a 10,5 meses, contra 9,1 meses (0,6 meses) de placebo más docetaxel, con un retraso en la progresión de la enfermedad de 4,5 a 3,0 meses (0.5 de diferencia). Resulta claro el ejemplo de una droga de muy alto precio con un efecto terapéutico comparativo notablemente pequeño.
Otro es el regorafenib (Stivarga®) de Bayer, un inhibidor multiquinasa administrable por vía oral para el tratamiento del cáncer de colon metastásico, su uso prolonga la vida 1,4 meses. Tiene un valor en México de U$ 9.350 para un ciclo de tratamiento de 28 días y en EEUU de entre U$ 12.000 a 14.000 (U$ 167/comprimido). Un editorial extraído de The Lancet señala que es poco probable calcular su futura rentabilidad, dada la modesta eficacia que posee y su elevada toxicidad.
El problema mayor se da con la competencia de los monoclonales o su combinación en busca de mayor efectividad frente a las químicas habituales y los valores de mercado. Si bien en las Fases clínicas pueden tener datos estadísticamente significativos de supervivencia libre de avance de enfermedad (SSP), su costo solo o en combinación puede resultar en cifras astronómicas.
En el año 2009 la FDA aprobó el uso de gefitinib (Iressa®) de Astra Zeneca por vía oral diaria como tratamiento de primera línea para el carcinoma de pulmón no microcitico metastasico con una mutación del receptor del Factor de crecimiento epidermal (EGFR) que provoca un evento antiapoptótico. Su acción consiste en inhibir la actividad de los caminos intracelulares implicados en el crecimiento y sobrevida de las células neoplásicas, a nivel del bolsillo de unión del dominio catalítico intracelular del receptor. El IPASS (IRESSA Pan-Asia Study) fue el primero de cuatro ensayos de fase III sobre 1.207 pacientes destinado a evidenciar superioridad confirmada. Posteriormente, el estudio IFUS efectuado sobre 106 pacientes naive de tratamiento mostró una media de sobrevida de 10.9 meses contra 7,4 meses para el grupo testigo tratado con carboplatino/paclitaxel (3.5 meses de diferencia).
Astra Zeneca ya había desarrollado en 2003 un primer estudio sobre 1.700 pacientes no elegidos. Los tratados con Iressa® vivieron un promedio de 5.6 meses contra 5.1 meses del grupo placebo, con lo cual la sobrevida se limitaba a dos semanas. Por tal motivo, en 2005 se lo retiro de comercialización en países occidentales. No obstante, en asiáticos la respuesta fue mejor, ya que mientras el grupo tratado con Iressa® vivió en promedio 9.5 meses, los bajo placebo solo lo hicieron durante 5.5 meses. La diferencia de sobrevida libre de progresión de enfermedad (PFS) fue así de 4 meses promedio.
El gefitinib es un genérico elaborado por 12 compañías a un precio promedio de entre U$ 319 a 400/mes y una duración media de tratamiento de 9.8 meses. A partir de su nueva indicación, con el nombre comercial de Iressa® paso a costar en presentación de 30 comprimidos alrededor de U$ 7.695 en EEUU y U$ 3.000 en Canadá. Su competencia es el nivolumab (Opdivo®) de BMS. Originalmente indicado para reducir el tamaño y prolongar la vida de los pacientes con melanoma avanzado, es un monoclonal inhibidor anti PD-1, proteína que impide que las células T reconozcan y ataquen los tejidos inflamados y las células cancerosas. La PD-1 puede engañar al sistema y lograr que las células del melanoma parezcan normales. LA AEM también ha autorizado su uso en la Unión Europea para tratamiento del carcinoma de pulmón avanzado previamente tratado con quimioterapia. El nivolumab mejora la sobrevida media 9,2 meses comparado con doxetacel (6.0 meses). Después de 12 meses de tratamiento, el 42 de los pacientes está vivo, comparado con el 24% de la serie con docetaxel. El problema vuelve a ser el precio. El nivolumab cuesta $ 28.78 por mg de la droga, por lo que un tratamiento completo implica un costo total de U$103,220 (para obtener una PFS de 6.9 meses).
En caso de usarse en el tratamiento del melanoma metastásico o no resecable, el costo del nivolumab puede ser extremadamente elevado por paciente, si se lo combina con otro monoclonal de nombre ipilimumab (Yervoy®) también de BMS y cuyo precio es de U$157.46 por mg. Un trial efectuado para demostrar efectividad de dicha combinación estableció que el costo de usar ipilimumab solo era de U$ 158.282 (para una mediana de supervivencia libre de avance [PFS] de 2,9 meses), el del nivolumab de U$ 103.220 (para una PFS de 6,9 meses) y la forma combinada de U$295.566 (PFS de 11,4 meses), casi cuatro veces la observada con ipilimumab solo, que es actualmente el estándar de cuidado. El principal inconveniente de la combinación es que produce graves efectos secundarios, como inflamación del colon, diarrea y problemas con las glándulas endocrinas.
Otra combinación de drogas para el cáncer de mama recientemente metastásico sin tratamiento previo o con recaída después de haber recibido tratamiento adyuvante o neoadyuvante, y en el que las células tumorales sobre-expresan la proteína HER2 (HER2 positivo), incluye el pertuzumab (Perjeta®) de Roche – Genentech. Su precio es de U$ 5.900 por mes, y es frecuente su utilización junto al trastuzumab (Herceptin®) de la misma compañía. Ambos fármacos sumados tiene un costo de U$ 10.400/mes, con una extensión promedio de tratamiento de 18,5 meses, lo que demuestra cómo las futuras asociaciones de drogas seguirán incrementando los costos a medida que más protocolos puedan incorporarse al mercado sanitario.
Las farmacéuticas discurren por una fina línea entre la poca efectividad en la sobrevida que otorgan las nuevas drogas oncológicas, el continuo incremento de sus precios y el nivel de indignación moral de la sociedad. El valor que agreguen en términos de beneficios se transforma en el nuevo mantra de la industria, lo que trata de definir a través de diferentes indicadores: beneficios respecto de supervivencia, progresión del tumor, mejora de la calidad de vida, reducción de la toxicidad asociada y de medicación concomitante y estadías hospitalarias más breves. Pero a medida que más innovaciones oncológicas llegan al mercado y más combinaciones de alto costo se vuelven comunes, el valor del medicamento oncológico se vuelve una asignatura cada vez más desafiante. Un dato a favor de la industria es que una de las razones por las cuales el costo global del tratamiento del cáncer está aumentando es porque los pacientes están siendo tratados durante un tiempo más prolongado que hace años. Y si la terapia está durando más, significa que los pacientes están sobreviviendo más tiempo. Pero esto no justifica bajo ningún concepto la disparada de precios.
Si la prevención viene primero que la atención. ¿Porque en oncología resulta un término casi ausente?
¿Venimos perdiendo la guerra contra el cáncer? Una pregunta necesaria. A principios del siglo XX, una persona de cada veinte podía tener cáncer. En la década de 1940 la proporción era de una cada dieciséis. Para la década de 1970 se redujo a una de cada diez. Hoy es una de cada tres personas quienes lo padecerán en el curso de su vida. Para 2014, se estimó un total de 1.665.540 nuevos casos de cáncer diagnosticados y 585.720 muertes solo en los EE.UU. El National Cancer Institute (NCI) admitió que los gastos médicos por atención del cáncer habían alcanzado los U$ 125 billones en 2013, con una proyección incremental a 2020 del 39% (U$ 173 billones).
Quizás sea esta una de los motivos de la frase del Dr. Linus Pauling, Premio Nobel en 1986, quien sugirió que «todo el mundo debe saber que la investigación sobre el cáncer es en gran parte un fraude.» Lo hizo con el conocimiento incipiente que la industria de lo oncológico se estaba volviendo un negocio rentable en los Estados Unidos, pero sin saber que el promedio de sobrevida provisto por los medicamentos aprobados entre 2002 y 2014 sería solo de 2.1 meses.
Según el IMS Institute for Healthcare Informatics, un nuevo tratamiento oncológico ingresado al mercado sanitario en 2002 tenía un costo promedio de U$ 4.500/mes. En la actualidad, el precio medio oscila en los U$ 10.000, y dos de las nuevas drogas aprobadas en 2014 ya superaron los U$ 35.000 por mes de tratamiento. Mientras tanto, el listado de innovaciones, sobre todo biotecnológicas, y las Fases de investigación continúa creciendo. Cincuenta de las nuevas drogas aprobadas en 2013 incluyen diecisiete destinadas a tratamiento oncológico, con un crecimiento en ventas de U$53 billones. Por ejemplo, los monoclonales rituximab (Rituxan®), bevacizumab (Avastin®) y trastuzumab (Herceptin®), todos del laboratorio Roche Genentech, obtuvieron ganancias por ventas combinadas cercanas a los U$21 billones, 40 % del valor de los 20 productos de mayor venta en el mercado sanitario. Junto a sus otras drogas erlotinib (Tarceva®) para el Cáncer de páncreas y capecitabina (Xeloda®) para el Cáncer de colon metastasico, gástrico y de mama, Roche generó por ventas totales en la banda terapéutica de oncológicas en 2013 cerca de U$ 24 billones. En 2010, Novartis recaudo, solo por ventas con imatinib (Gleevec®), U$ 4.3 billones.
Si la dinámica de la industria farmacéutica innovadora es la búsqueda rápida de tratamientos para fases cada vez más avanzadas de la enfermedad ¿Dónde ha quedado la investigación en prevención especifica del cáncer? Ciertos estudios han demostrado que un uso regular y prolongado de ácido acetil salicílico (Aspirina®) ha demostrado ser útil para reducir el riesgo de adenomas, cáncer colorrectal y su forma recurrente en seres humanos, aunque en otros tumores dicha utilidad ha resultado inconsistente. Tanto la Aspirina® como los Analgésicos No Esteroides (AINE) como el rofecoxib pueden reducir el riesgo de cáncer aún no está claro, pero parece estar ligado a su capacidad para bloquear la enzima ciclooxigenasa (COX). También el tamoxifeno, aprobado por la FDA como el primer agente quimioprotector para mujeres con alto riesgo de cáncer de mama muestra datos estadísticamente significativos.
Recientemente, la División de Prevención del Cáncer del National Cancer Institute (NCI) ha identificado dos áreas prioritarias dentro del Programa PREVENT de intervenciones y biomarcadores destinadas al desarrollo preclínico de drogas. Estas dos áreas clave pueden favorecer un elevado retorno de la inversión a las empresas mediante ensayos específicos durante un período relativamente corto de tiempo. La primera pasa por el desarrollo de antiinflamatorios específicamente dirigidos a la prevención del cáncer. Definir biomarcadores potenciales relacionados con el bloqueo de las vías de inflamación sería una vía de investigación. Este enfoque aparece como particularmente prometedor dado expresado respecto de la eficacia observada con los AINE y coxibs en estudios preclínicos y clínicos. La segunda se centra en la prevención inmune.
El desarrollo de vacunas contra el cáncer en etapas tempranas también aparece como una alternativa factible en estudios preclínicos. Existe una cantidad sustancial de datos recientes en relación al uso de vacunas o de células T activadas en un entorno terapéutico. Este segundo enfoque ofrecería una excelente estrategia para combatir el proceso de cáncer temprano con toxicidad mínima. En momentos en que el número creciente de medicamentos cada vez más costosos pone en duda la sustentabilidad de los sistemas de salud, especialmente de los países con menores ingresos, la investigación en prevención sobre el cáncer se vuelve imprescindible. Los institutos de salud de Estados Unidos han admitido la existencia de más de 1.000 drogas oncológicas en fase de desarrollo. Cerca de 123 están dirigidas a tratar el cáncer de pulmón, principal causa de muerte. 106 corresponden a terapias para formas severas de leucemias, 92 para linfomas incluyendo la forma no Hodking, 82 para cáncer de mama, 58 para tumores cerebrales (incluyendo el glioma que representa 80% de todos los tumores malignos) y 53 para cáncer de piel (incluyendo al melanoma altamente agresivo)Pero la mayor parte de los costos de investigación no pertenecen a la industria, sino a gobiernos y consumidores que financian el 84% de la investigación aplicada, en tanto solo 12 % corresponde a los laboratorios. Este dato hace caer el mito del precio de las drogas atado a la inversión en I+D. Un estudio efectuado sobre 117 protocolos de investigación demostró que el promedio de inversión privada solo ronda en los U$80 millones. La FDA admite que solo 20% de la inversión en investigación corresponde a productos que aportan innovación y mejora terapéutica significativa comparada con otras drogas ya existentes en el mercado. Es la condición de monopolio establecida por la patente lo que permite a las empresas fijar arbitrariamente los precios, muy lejos de los costos de producción. Paradójicamente, el 75% del financiamiento de dicha agencia regulatoria proviene de la propia industria a quien debe regular.
Conclusiones
La industria farmacéutica apunta a la innovación permanente. Y las patentes de medicamentos son su principal incentivo. Especialmente con las biotecnológicas, cuestión ya incorporada específicamente al Tratado Asia Pacifico. La aprobación de una nueva droga por la FDA es un cheque en blanco en relación con la seguridad de esta y su eficacia. Posteriormente, la fase de farmacovigilancia permite ajustar los problemas surgidos a lo largo de su uso en el mercado.
Durante la primera década del siglo XXI, se han dado pasos importantes en la comprensión de la biología del cáncer en general y, por tanto, la terapia específica. Un paso importante fue la comprensión de la variación de fenotipo / genotipo celular intratumoral, que podría explicar las diferencias de eficacia de la droga en términos de mejora clínica y ganancias de bienestar. Lo mismo para la identificación de una serie de procesos que se producen en la interacción dé las células tumorales con su microambiente, que gobiernan la evolución de la metástasis. La biotecnología confirma que, para cada paciente, existe una secuenciación de genes, patrones de señalización intracelular, grupos de biomarcadores y dianas terapéuticas que allanan el camino hacia la medicina personalizada y la target therapy sobre el paciente. Precisamente, los investigadores están en la búsqueda de biomarcadores que expresen diversos estados de la enfermedad. Puede tratarse de proteínas cuyos niveles se correlacionen con predecir el riesgo de enfermedad, diagnosticarla precozmente o utilizarlas como indicadores de resultado del curso de tratamiento.
Los investigadores están explorando nuevos métodos de alta tecnología para librar una batalla más pareja contra la enfermedad, así como nuevas formas de maximizar el uso de los medicamentos existentes, sea solos o en combinación con otras terapias. De hecho, aproximadamente 80% de las drogas oncológicas en la pipeline son medicamentos first in class y 70% se perfilan para formar parte de la medicina personalizada. De cerca de 836 drogas actualmente bajo desarrollo, El mayor inconveniente en la búsqueda de tratamientos para prevención y/o atención de determinadas formas de cáncer radica en la distorsión que produce sobre la industria farmacéutica el tipo de incentivos, que condicionan el desarrollo de determinadas líneas de tratamiento. De la misma forma actúan los elevados precios de mercado conque introducen sus productos y la respuesta de la competencia y también del uso que se le da a ciertas drogas desde lo asistencial y el valor que eso significa en términos de ganancia de vida. Pero fundamentalmente es la elasticidad/precio el factor que la industria encuentra tanto en el momento del lanzamiento de un nuevo producto como en los posteriores incrementos de precios, que no muestran signos de detenerse.
Muchos aseguradores a nivel mundial, como el caso de Express Scripts Holding Co., una gerencia dora de beneficios de prescripción de drogas en EEUU, comienzan a plantear la posibilidad de cerrar acuerdos con empresas farmacéuticas para pagar menos cuando las drogas a utilizar, de muy alto precio, no generan frente a determinado tipo de tumores e indicaciones, los resultados esperados. Un mecanismo denominado pago por perfomance.
El caso del erlotinib sirve de testigo sobre la diferencia en efectividad y precio según la indicación, tipo de tumor y estadio de avance. Administrado por vía oral en el caso de cáncer pancreático no resecable o metastàsico, la sobrevida media que obtiene frente a placebo no supera las dos semanas. En cáncer de pulmón, su efectividad es de 6.7 meses contra 4.7 meses de una combinación de quimioterapia tradicional. En base a estas amplias diferencias, el precio por comprimido de la droga debería ser inferior para el primer caso que para el segundo. El inconveniente es que la droga tiene un precio único, para cualquiera de las dos alternativas, de U$ 7.224 los 30 comprimidos de 150mg.
Hoy existe una amplia variedad de tratamientos que prolongan la vida de pacientes con tumores metastásicos o avanzados. También muchas de las nuevas drogas se han convertido en terapia común para pacientes con enfermedad en estadios tempranos, post cirugía o radioterapia, empleándolas como adyuvantes. Especialmente aquellas con marcadores genéticos. Estas terapias dirigidas tienen más probabilidades de resultar exitosas a lo largo de los ensayos clínicos. El dilema surge entre el conocimiento científico encarnado por los nuevos fármacos, especialmente los biotecnológicos, y el progreso en términos de alternativas terapéuticas disponibles, cuando no se acompaña de mejoras proporcionales en los resultados que obtienen los pacientes en términos de beneficios netos. Las ganancias en Años de Vida Ajustados por Calidad, desafortunadamente, se siguen midiendo en oncología en semanas o meses, y no en años.
Es por esa razón que las drogas destinadas a pacientes poseedores de una esperanza de vida corta pueden moverse con más rapidez a lo largo de los ensayos clínicos que otras destinadas a quienes poseen mayor expectativa de vida. Pero esto no lleva a que los elevadísimos precios de los medicamentos se modifiquen a la baja después de su lanzamiento. Precisamente porque ese es el punto de inflexión de la industria. ¿Cómo se deciden los precios de los medicamentos contra el cáncer? De los muchos factores complejos que parecieran estar implicados, parece seguirse una fórmula sencilla: situar el precio más reciente de una droga similar en el mercado y sumarle de base un 10 a 20%. Aunque los oncólogos no enfrentan incentivos directos para evitar las drogas más costosas, pueden resistirse a la prescripción de aquellos medicamentos cuyos precios se encuentren por encima del nivel de precios de referencia. Existe una «zona de indiferencia» en torno a un precio de referencia, que les permite a los consumidores ignorar las desviaciones del precio final respecto de este, y a los fabricantes otorgarles la posibilidad de fijar precios excesivamente altos a nuevos medicamentos, sin que se reduzca la cantidad demandada.
La estructura del mercado de medicamentos, en este caso para terapia oncológica, incluye la protección de las patentes y no ofrece otros mecanismos de control de precios que no surjan de la buena voluntad de financiadores y pacientes y de la tolerancia de las empresas a la publicidad adversa respecto de conductas monopólicas y abusivas y de que los aumentos no guardan relación con la magnitud de los beneficios esperados para la salud. El proceso por el cual las empresas establecen los «precios de lanzamiento» de nuevas drogas resulta bastante opaco, y en el momento de la aprobación de comercialización por la FDA, la mayoría de estas ya son de patente, por lo cual la industria constituye monopolios temporales con amplia libertad de acción e ilimitado poder para fijar los precios.
La terapia oncológica se hace cada vez más compleja y acelera el desarrollo de nuevos productos. Estamos lejos de la prevención. Los precios de las drogas suben por el ascensor y los financiadores sienten temblar sus estructuras económicas. Los reguladores se sienten regulados y los médicos le escapan a los protocolos en nombre de la autonomía profesional. Y los pacientes reciben miles de tratamientos, muchos experimentales, con baja evidencia de beneficios en calidad o cantidad de vida efectiva. La encrucijada se transforma en dilema. El problema, como la Alicia del cuento, es qué camino tomar. La farmacoeconomía puede ayudar, pero necesita de la política sanitaria. Como escribí hace no mucho, parafraseando al gran Leonardo “La técnica sin la política es manca, pero la política sin la técnica es ciega”.
Bibliografía
- Frakt, Austin. Why Preventing Cancer Is Not the Priority in Drug Development. The New York Times. DEC. 28, 2015. Encontrado en: http://www.nytimes.com/2015/12/29/upshot/whypreventing-cancer-is-not-the-priority-in-drug-development.html?partner=rss&emc=rss&_r=0
- Eric Budish, Benjamin N. Roin, and Williams, H. Do Firms Underinvest in Long-Term Research? Evidence from Cancer Clinical Trials. American Economic Review 2015, 105(7): 2044–2085 http://pubs.aeaweb.org/doi/pdfplus/10.1257/aer.20131176
- Chambers J.D., Thorat T, Pyo,J, Chenoweth M, Neumann P; Despite high costs, specialty drugs may offer value for money comparable to that of traditional drugs. Health Aff (Millwood). 2014 ,Oct;33 (10);:1751-60. Encontrado en: http://www.natap.org/2014/HCV/HealthAff–2014–Chambers–1751–60.pdf
- Siddiqui,M &Rajkumar, S.V; The High Cost of Cancer Drugs and What We Can Do About It.
- Mayo Clinic Proceedings; Volume 87, Issue 10, Pages 935–943: http://www.mayoclinicproceedings.org/article/S0025-6196(12)00738-0/abstract
- Jacobson M, Earle CC, Price M, Newhouse JP. How Medicare’s payment cuts for cancer chemotherapy drugs changed patterns of treatment Health Aff (Millwood). 2010 Jul; 29 (7):1391-9. http://content.healthaffairs.org/content/29/7/1391.abstract
- Sassi, F. Calculating QALYs, comparing QALY and DALY calculations. Health Policy Plan.2006 ;21: 402–408.
- The Patient Protection and Affordable Care Act(PL 111-148) . ; March 23, 2010.
- Longo,D; Cancer-Drug Discovery — Let’s Get Ready for the Next Period; N Engl J Med 2014; 371:2227-2228; http://www.nejm.org/doi/full/10.1056/NEJMe1412624
- Meropol, N.J., Schulman, K.A. Cost of cancer care: issues and implications. J Clin Oncol.2007; 25:180–186. http://jco.ascopubs.org/content/25/2/180.full.pdf+html
- Karnon,J & col. New cancer drugs are very expensive – here’s how we work out value for our money; September 8, 2015 http://theconversation.com/new–cancer–drugs–are–veryexpensive–heres–how–we–work–out–value–for–our–money–44014
- DiMasi, J.A., Grabowski, H.G. Economics of new oncology drug development. J Clin Oncol.2007; 25:209–216. http://jco.ascopubs.org/content/25/2/209
- Kelloff,GJ& Sigman;CC New science-based endpoints to accelerate oncology drug development.
- Eur J Cancer; 2005 Mar; 41 (4);491-501. http://www.ncbi.nlm.nih.gov/pubmed/15737552
- FirstWordPharma. US cancer doctors drop pricey drugs with little or no effect; October 2015; http://www.firstwordpharma.com/node/1321368?tsid=33#axzz3yY9NQVqk
- McKee, A.E., Farrell, A.T., Pazdur, R., Woodcock, J. The role of the U.S. Food and Drug Administration review process: clinical trial endpoints in oncology. The Oncologist. 2010; 15:13–18. http://theoncologist.alphamedpress.org/content/15/suppl_1/13
- David H. Howard, Peter B. Bach, Ernst R. Berndt, and Rena M. Conti; Pricing in the Market for Anticancer Drugs; Journal of Economic Perspectives—Volume 29, Number 1—Winter 2015—Pages 139–162. Encontrado en: http://pubs.aeaweb.org/doi/pdfplus/10.1257/jep.29.1.139
- Shaughnessy, A; Monoclonal antibodies: magic bullets with a hefty price tag BMJ 2012; 345 doi: http://dx.doi.org/10.1136/bmj.e8346;
- Mok TS et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Eng J Med 2009; 361. 10.1056/NEJMoa0810699. http://www.nejm.org/toc/nejm/361/10
- Chabner,B; The Miracle of Iressa®;The Oncologist: Journal of the Society for Traslacional Oncology; January 2016 http://theoncologist.alphamedpress.org/content/9/3/245.full
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al: Efficacy and safety results from a phase III trial of nivolumab alone or combined with ipilimumab versus ipilimumab alone in treatment-naive patients with advanced melanoma. 2015 ASCO Annual Meeting. Abstract LBA1. Presented May 31, 2015. http://meetinglibrary.asco.org/content/144621–156
- Schnipper; L & Meropol, N; Payment for Cancer Care: Time for a New Prescription. Journal of Clinical Oncology; Volume 32 Nro 36 December 20 2014 http://jco.ascopubs.org/content/32/36/4027.full.pdf+html
